PO:22:039 | A rare form of interstitial lung disease: granulomatous and lymphocytic interstitial lung disease
Francesca Cozzini1|2, Francesco Falco1, Alberto Cavazza1, Lucia Spaggiari3, Chiara Marvisi1, Francesca Petrillo1|2, Caterina Ricordi1|2, Francesco Muratore1|2, Fabio Brandolino1, Ilaria Chiapparoli1, Carlo Salvarani1|2, Andreina Manfredi1|2 | 1IRCCS Policlinico San Matteo, UO di Reumatologia, Pavia; 2Università degli studi di Modena e Reggio Emilia, Modena, Italy
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Authors
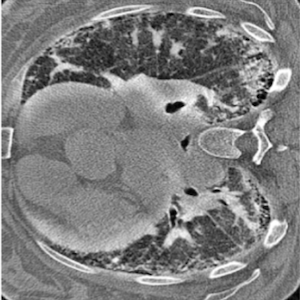
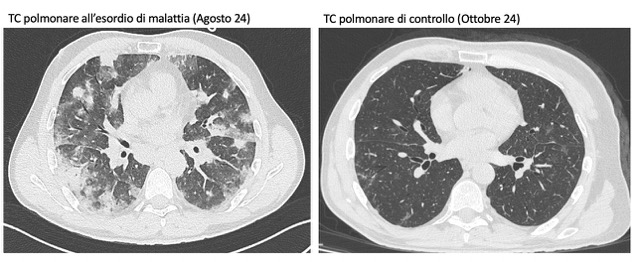
Background. In August 2024, a 48-year-old man presented to the emergency department with worsening dyspnea and oxygen desaturation. His medical history was remarkable only for IgA deficiency, but he reported recurrent episodes of pneumonia over the previous two months, treated with multiple antibiotics without improvement. He was admitted to the Department of Pulmonology at our hospital because his chest X-ray showed pulmonary infiltrates, his inflammatory markers were high, and he needed oxygen therapy. Chest CT performed during hospitalization showed diffuse nodular consolidations and ground-glass opacities predominantly in the middle and lower pulmonary lobes bilaterally. Autoimmune screening (ANA, ENA, ANCA, RF, anti-CCP, and myositis blot), serum precipitins, and microbiological tests (blood cultures, galactomannan, β-D-glucan, CMV DNA, and Borrelia serology) were all negative. Rheumatologic evaluation revealed no signs or symptoms suggestive of connective tissue disease. During hospitalization, treatment with iv methylprednisolone 80 mg led to oxygen therapy withdrawal and improvement of pulmonary consolidations on chest CT. The patient was discharged with a diagnosis of organizing pneumonia and prescribed prednisone 50 mg daily with gradual tapering. In January 2025, a new pulmonary consolidation was observed on CT scan. To further clarify the clinical picture, the patient underwent bronchoalveolar lavage (showing 50% lymphocytes, 30% macrophages) and transbronchial biopsy. Histopathological examination revealed foci of organizing pneumonia, lymphocytic inflammation, and nonnecrotizing granulomas, findings suggestive of Granulomatous and Lymphocytic Interstitial Lung Disease (GLILD). The case was discussed at the multidisciplinary interstitial lung disease meeting (including radiologists, pulmonologists, pathologists, rheumatologists), where a diagnosis of GLILD secondary to IgA deficiency was confirmed. The patient began treatment with Rituximab 1000 mg, which was given twice, two weeks apart. At follow-up, the patient reported sustained clinical well-being and had resumed his work activities.
Discussion. GLILD is a rare interstitial lung disease secondary to common variable immunodeficiency or selective IgA or IgG deficiency. The onset is often acute, presenting with cough and dyspnea, although asymptomatic cases have been described. According to literature data, high-resolution chest CT is the first-line imaging modality, typically showing peribronchovascular pulmonary nodules, ground-glass opacities, and reticulations in the lower lobes. Lung biopsy is the gold standard for diagnosis, usually revealing peribronchial lymphocytic infiltrates, non-necrotizing granulomas, follicular bronchiolitis, and foci of organizing pneumonia. Treatment generally consists of corticosteroids, while rituximab is indicated in refractory cases. There is no clear evidence supporting the efficacy of intravenous immunoglobulin replacement therapy in GLILD.
Conclusions. GLILD is a rare autoimmune-associated interstitial lung disease with no established treatment guidelines. Therefore, a multidisciplinary approach is crucial for accurate diagnosis and optimal therapy.

Downloads
Citations




How to Cite

This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License.